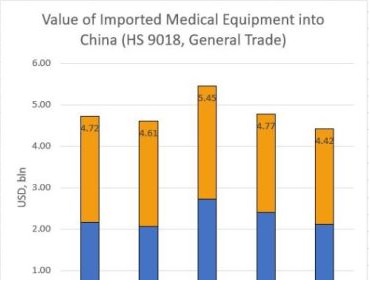
Medical Device Registration Procedure in China.
China Medical Device Regulatory Body
Everyone interested in selling medical products in China needs to register them. The regulatory body responsible for this process is called the National Medical Products Administration (NMPA), in Chinese 国家药品监督管理局. However, NMPA is more like a registrar that keeps track of records about the products with valid and not-valid medical device registration certificates. The main work related to medical device evaluation is done by the Center for Medical Device Evaluation (CMDE), in Chinese 中国药品监督管理局医疗器械技术审评中心.
Medical Device Categories
China has three categories of medical devices: Class I, Class II and Class III.
Class I medical devices refer to medical equipment for which routine management is sufficient to ensure its safety and effectiveness, such as surgical instruments and medical dressings.
Class II medical devices are those for which control of safety and effectiveness is necessary, such as physical therapy and rehabilitation equipment, clinical diagnostic analyzers, etc.
Class III medical devices are those that are implanted in the human body to support or sustain life and pose potential risks to the human body. Strict control of their safety and effectiveness is required. Examples include disposable, sterile medical devices, and implantable devices.
When determining the product category, one can refer to the classification catalog by directly searching for the standard product name. If the product name cannot be determined, the category can be determined based on the product description and intended use.

Product Testing
Before the registration of medical devices, it is necessary to verify the product's parameters and obtain a testing report from an organization with the appropriate qualification for review. For Class I medical device products, the testing report is issued by the manufacturing company itself. However, for Class I products that manufacturers cannot test themselves, it is necessary to entrust a testing organization with the testing process. For Class II and Class III medical device products, testing must be entrusted to nationally accredited testing organizations.
Companies are required to provide product technical specifications, and testing personnel determine the product's compliance with the parameters specified in the technical requirements. Typically, the testing is carried out by one of the 53 medical device testing institutes under the supervision of the Drug Administration. These institutes issue preliminary assessments and registration testing reports to the applicants for medical devices that meet the requirements.
After meeting the testing requirements, active medical devices need to provide electromagnetic compatibility reports and comprehensive testing reports, whereas passive medical devices only need to provide comprehensive testing reports. It's important to note that the presence of the CMA (China Metrology Accreditation) logo on the report cover is a focal point for regulatory personnel during the registration process.
The production of registered inspection samples must conform to the relevant requirements of the medical device quality management system. Only after passing the registration inspection can clinical trials be conducted, and products that are exempt from clinical trials can proceed to the system establishment and registration phase.

Clinical Trials
Regarding clinical trials, there are several situations in which clinical trials are not required. Class I medical device products are considered low-risk and, therefore, do not need to undergo clinical trials. On the other hand, Class II and Class III medical device products that are not included in the national medical device exemption catalog or cannot obtain clinical evaluation data through comparison with similar products require clinical trials.
Before conducting clinical trials, it is necessary to obtain clinical trial registration from the regulatory authority. Then, suitable clinical trial institutions need to be selected, and a detailed clinical trial protocol needs to be developed. The protocol undergoes review during an ethics committee meeting to ensure its feasibility. Once the protocol is deemed executable, formal clinical trials can commence.
Clinical trials require the recruitment of a sufficient number of subjects to ensure the accuracy and reliability of the results. Throughout the trial, clinical coordinators and clinical monitors closely oversee the process to ensure the orderly progress of the project.
Upon completion of the project, a clinical evaluation report is issued to summarize and assess the trial results. At this point, the clinical trial phase officially concludes.
For Class II and Class III medical devices and IVD (In Vitro Diagnostic) products, the specific determination of the sample size is calculated based on relevant formulas. Typically, the sample size for Class II and Class III medical devices is around 140 cases, while the sample size for Class II and Class III IVD products may require several hundred or even several thousand cases, depending on the circumstances.
The medical device registration process in China requires product testing, clinical trials and application (only by China registered company). Class I medical devices do not require clinical trials. Some class II and class III medical devices may also be exempt from clinical trials.
Application
In theory, once clinical trials are completed, the product registration documents can be submitted. The applicant should submit the application materials to the National Medical Products Administration in accordance with relevant requirements. The NMPA department responsible for accepting the registration application should transfer the application materials to the Center for Medical Device Evaluation within 3 working days from the date of acceptance. CMDE should complete the registration technical evaluation for Class II medical devices within 60 working days and for Class III medical devices within 90 working days. If the registration materials do not pass evaluation, the regulatory authority will issue a request for supplementary materials, and the applicant must complete the required corrections within the specified time frame and resubmit. Only after passing this stage can the process proceed to the next phase.
NMPA department responsible for accepting registration applications shall decide within 20 working days after the completion of the technical evaluation process. If the applied product meets the safety and efficacy requirements, it will be granted registration approval. Within 10 working days after making the approval decision, NMPA will issue a medical device registration certificate to the applicant and send the approved product's technical requirements as an attachment to the applicant.
If the registration application is denied, the regulatory authority shall provide written reasons for the refusal and inform the applicant of their right to apply for a reexamination and to seek administrative reconsideration or initiate administrative litigation in accordance with the law.
Sources
Article I
Article II
Related Posts